
Total de visitas: 337682
|
Engenharia Genética
O que é? Engenharia Genética é o termo utilizado para descrever algumas técnicas modernas que têm vindo a revolucionar o campo da Biotecnologia. Consiste num conjunto de técnicas e ferramentas que permite identificar, isolar, manipular e multiplicar os genes de organismos vivos. Estas técnicas incluem a cultura de células, o uso de isótopos radioactivos, a clonagem molecular, a tecnologia do ADN (ácido desoxirribonucleico) recombinante e o aperfeiçoamento das técnicas de obtenção, purificação e manipulação de enzimas. O objectivo é introduzir novas características ou atributos fisiológicos ou físicos em seres vivos, como por exemplo, tornar uma planta resistente a um herbicida.
Deve ter-se em conta que isto não é Engenharia no sentido convencional. O termo Engenharia geralmente implica um total conhecimento dos resultados de uma dada intervenção. Contudo, há muitos exemplos de alterações de sequências de ADN num organismo que levam a alterações sistémicas muitas vezes inesperadas.
História Os pesquisadores norte-americanos George W. Beadle e Edward L. Tatum, na década de 1930, demonstraram que a produção de proteínas e enzimas era regulada pelos genes e que, consequentemente, estes últimos intervinham nas múltiplas reacções que ocorrem nos seres vivos. Destas pesquisas, resultou o processo de descoberta da estrutura genética do Homem.
Oswald T. Avery em 1944, ao analisar a cadeia polinucleotídica doDNA, descobriu que este é o componente cromossómico que permite a transmissão das informações genéticas.
Em 1953, o inglês Francis H. C. Crick e o norte-americano James D. Watson, com base nos trabalhos de Chargaff, Rosalind Franklin e Maurice Wilkins, apresentaram o modelo de dupla hélice para a molécula de DNA.
Em 1961, os franceses François Jacob e Jacques Monod estudaram o processo de síntese de proteínas nas células bacterianas. Descobriram que o responsável pela regulação desta síntese era o DNA, que passou então a ser o elemento central das pesquisas de engenharia genética.
Em 1972, na Universidade de Stanford, na Califórnia, o norte-americano Paul Berg ligou duas cadeias de DNA, sendo que uma era de origem animal e a outra de origem bacteriana. Esta foi a primeira experiência bem sucedida onde foram ligadas duas cadeias genéticas diferentes e é considerada por muitos autores o início da criação sintética de produtos de engenharia genética.
Em 1978, o suíço Werner Arber e os norte-americanos Daniel Nathans e Hamilton O. Smith receberam com o Prémio Nobel da Medicina por terem isolado as enzimas de restrição (enzimas capazes de seleccionar e cortar o DNA em locais específicos desta molécula).
Iniciou-se então a era da manipulação genética, consistindo, de uma forma muito global, na manipulação de mensagens genéticas expressas em fragmentos de sequências que compõem o código hereditário, os nucleótidos.
As técnicas Seguidamente são apresentadas as técnicas fundamentais pertencentes ao domínio da Engenharia Genética. São elas a técnica do DNA recombinante (rDNA), a técnica do DNA complementar (cDNA), que se inclui na técnica do DNA recombinante, a técnica da Reacção de Polimerização em Cadeia (PCR) e a tecnologia das Impressões Digitais Genéticas ou DNA fingerprinting.
DNA recombinante (rDNA)
Esta tecnologia tem como objectivo produzir moléculas de DNA a partir da combinação de genes com proveniências diferentes.
Primeiramente, é necessário obter os genes para o enxerto. Existem três formas fundamentais de obtenção destes genes:
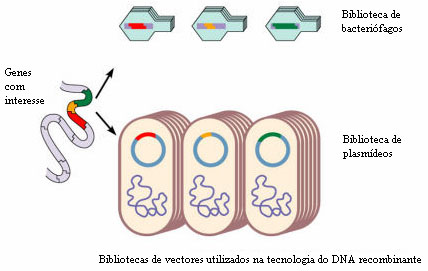
1. Criar-se uma biblioteca de genes ou banco genómico, que consiste numa colecção de um grande número de fragmentos de DNA do genoma que são armazenados em vectores (de que falaremos adiante) para posteriores utilizações;
2. Fabricar-se o gene em laboratório a partir de RNA mensageiro (mRNA) (técnica do DNA complementar de que se falará posteriormente);
3. Sintetizar-se o gene, nucleótido por nucleótido, na sequência desejada. Isto é feito normalmente para genes que codificam polipéptidos ou proteínas de pequeno tamanho. Para executar esta técnica basta conhecer a sequência de aminoácidos da proteína desejada e, com base no conhecimento do código genético, construir a sequência de nucleótidos que corresponde exactamente à sequência de aminoácidos da proteína.
Depois de se obter o gene de interesse, é necessário que enzimas de restrição cortem o DNA em fragmentos manipuláveis que contêm o gene específico pretendido. Estas enzimas, também conhecidas como endonucleases, são enzimas que clivam a molécula de DNA por hidrólise através do reconhecimento de sequências nucleotídicas específicas. Ocorrem naturalmente em bactérias (o DNA bacteriano está protegido da actividade das enzimas de restrição), protegendo-as dos ataques dos vírus, uma vez que reconhecem e cortam sequências específicas do DNA viral, inactivando-o. Contudo, actualmente, estas enzimas são produzidas por empresas de biotecnologia e são uma das ferramentas básicas em Biologia Molecular e Engenharia Genética.
Após a clivagem, formam-se fragmentos de DNA, em dupla hélice, mas com uma pequena extensão em cadeia simples em cada extremidade (extremidades coesivas). Estes pedaços de DNA designam-se fragmentos de restrição.

Os fragmentos de DNA que contêm os genes de interesse são, depois, incorporados numa entidade constituída por DNA capaz de transportar o fragmento de DNA para uma célula. Estas entidades designam-se vectores. Os bacteriófagos, vírus que atacam as bactérias, podem ser usados como vectores. Muitas bactérias, para além da cadeia de DNA principal, possuem cadeias de DNA livres e de forma circular, os plasmídeos, cujos genes, geralmente, não são essenciais à sobrevivência da bactéria, podendo ser retirados e usados também como vectores. Os plasmídeos podem replicar-se independentemente da molécula de DNA principal e podem também fundir-se com ela. Para além destes vectores mais conhecidos, são usados ainda os lipossomas. Estes são, no essencial, esferas formadas por camadas bilipídicas preenchidas com DNA. Fundem-se espontaneamente com a membrana celular, libertando o seu conteúdo no citoplasma. Estes vectores apresentam, contudo, pouca eficiência.

Desta forma, a mesma enzima de restrição que actuou no DNA com o gene de interesse, actua agora sobre o vector, cortando a cadeia de DNA deste em zonas específicas, expondo uma sequência nucleotídica complementar.
As extremidades coesivas do fragmento ligam-se, então, à cadeia exposta de DNA do vector, estabelecendo-se ligações de hidrogénio entre bases complementares. A enzima DNA ligase promove a formação de ligações fosfodiéster entre as extremidades coesivas de cada uma das moléculas de DNA.
Desta forma, é produzida uma molécula híbrida estável, o DNA recombinante.

Uma vez preparados os vectores é necessário inseri-los em células bacterianas. Assim, os vectores são postos em contacto com estas células, contudo, apenas algumas bactérias conseguem absorver o DNA novo, sendo necessário agora, seleccionar as bactérias que realmente incorporaram o DNA recombinante. Por exemplo, para facilitar a identificação das bactérias que incorporaram o DNA recombinante, os pesquisadores escolhem plasmídeos que possuem genes que conferem resistência a determinado antibiótico. Estes plasmídeos são colocados em contacto com bactérias sensíveis ao antibiótico, sendo que algumas destas os incorporam. Para seleccionar as bactérias que incorporaram o plasmídeo basta adicionar o antibiótico ao meio de cultivo. Todas as bactérias sem o plasmídeo morrem, restando somente as que possuem o plasmídeo com o gene para resistência ao antibiótico. Estas bactérias reproduzem-se e todos os clones resultantes possuirão o plasmídeo com o gene novo.
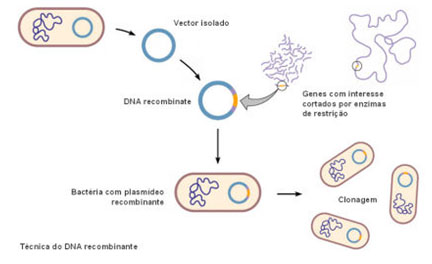
Assim, esta técnica permite, por exemplo, introduzir porções de DNA provenientes de vários organismos num microrganismo, que, em condições favoráveis, permite a produção de inúmeras cópias do gene (que poderão ser armazenadas nas bibliotecas de genes) ou da proteína codificada por este.
Resta falar, então, das mais-valias desta tecnologia, dando alguns exemplos das suas aplicações:
- Investigação fundamental esta técnica permite isolar genes de organismos complexos e estudar as suas funções a nível molecular.
- Obtenção de organismos geneticamente modificados (OGM). Os OGM ou transgénicos são organismos cujo genoma foi manipulado, apresentando diferenças relativamente à sua constituição original. Esta manipulação permite:
- Produção de alimentos em maior quantidade e qualidade:
- Resistência biológica a pragas e doenças específicas, reduzindo-se a necessidade de pesticidas químicos e diminuindo-se o risco de perdas nas plantações e aumentando a produção;
- Adaptabilidade a condições severas de crescimento, tais como seca, solo com alto teor de sal e temperaturas extremas. Por exemplo, modificando a produção de ácido linoleico de uma planta, esta pode resistir melhor a temperaturas baixas;
- Características funcionais desejáveis, como redução da alergenicidade ou toxicidade, retardo no amadurecimento, aumento do conteúdo de amido, ou uma durabilidade maior. Por exemplo, com o rDNA, batatas modificadas podem ter um conteúdo maior de amido, absorvendo menos óleo quando fritas, resultando em batatas fritas com menos gordura. Também tomates modificados de forma ao seu amadurecimento ser retardado podem permanecer no tomateiro por mais tempo, resultando num sabor e numa cor melhores antes de serem colhidos e enviados para o mercado;
- Características nutricionais desejáveis, como a alteração do conteúdo de proteínas ou gorduras. Problemas mundiais de desnutrição, como deficiência em vitamina A, ferro, iodo e zinco, podem ser combatidos pelo uso da tecnologia do rDNA, introduzindo ou concentrando estes nutrientes nas plantas. Por exemplo, o arroz tem sido geneticamente modificado para conter β-caroteno e mais ferro para ajudar a superar deficiências destes nutrientes em países onde o arroz é o alimento principal. Alimentos enriquecidos nutricionalmente podem até mesmo ajudar a prevenir doenças crónicas, por conterem níveis óptimos de nutrientes-chave.
- Produção de grandes quantidades de substâncias com aplicação farmacêutica ou médica Por exemplo, cientistas canadianos conseguiram transformar vesículas seminais de ratos em "biorreactores" (fábricas biológicas de substâncias de interesse). Para testar a viabilidade da técnica foi escolhida a proteína hGH (hormona de crescimento humano). Para montar o "gene artificial", além da sequência de bases contendo as instruções para produzir o hGH, foi utilizado também um promotor. O DNA construído foi injectado em vários embriões de ratos. Obteve-se então, animais transgénicos produzindo hGH no seu sémen. O próximo passo neste método é conseguir produzir a hormona de crescimento no fluído seminal do porco, dado que este é o animal doméstico que ejacula maior volume de líquido seminal.
- Produção de substâncias com aplicação industrial A manipulação genética de organismos pode ser posta em prática para produção de certas proteínas de interesse para a indústria têxtil. Um exemplo disto é proteína da seda produzida por bactérias geneticamente modificadas tendo como objectivo o melhoramento das propriedades da fibra. Também na produção de corantes são usados microrganismos que conseguem produzir pigmentos até cerca de 30% do seu peso seco e, consequentemente, a produção de corantes para tingimento de tecidos por via microbiana poderá vir a substituir a síntese química e assim evitar os inerentes problemas de libertação de produtos tóxicos para o meio ambiente.
- Biorremediação É uma das ferramentas da biotecnologia ambiental que utiliza microrganismos decompositores no tratamento dos resíduos, através da decomposição anaeróbia, bioestabilizando os resíduos, tornando-os menos solúveis, e portanto, menos perigosos. O rDNA é frequentemente utilizado para aumentar a concentração de microrganismos decompositores conferindo maior produtividade ao processo de tratamento.
- Tratamento de doenças Por exemplo, o rDNA permitiu reverter os efeitos de uma doença genética chamada imunodeficiência combinada grave ligada ao cromossoma X (SCID). Os indivíduos que sofrem desta doença são obrigados a viver em ambientes completamente isolados já que o seu sistema imunitário não defende o corpo de infecções. Num destes casos, a doença impede a produção de glóbulos brancos pela medula óssea. Assim, os pesquisadores retiram do vírus (vector) os genes que o tornam capaz de causar doenças. No lugar destes, insere-se o gene que produz, de forma correcta, a proteína que se encontra defeituosa no paciente. O vírus modificado é misturado com células da medula óssea retiradas dos pacientes, infectando-as. Com o gene terapêutico, as células passam a produzir a proteína responsável pela estimulação das células de defesa, fazendo com que estas se desenvolvam e se espalhem pelo corpo, destruindo os invasores.
Como se pôde constatar, através da tecnologia do rDNA são obtidos diversos produtos importantes para o Homem.

DNA complementar (cDNA)
O DNA complementar (cDNA) é um outro processo através do qual se podem conseguir bibliotecas de genes e é utilizado, na maior parte das vezes, como um auxiliar da técnica do DNA recombinante. O cDNA é obtido a partir do mRNA maturado (que já sofreu processamento e que, portanto, não possui intrões) por complementaridade de bases. Esta transcrição em sentido inverso é conseguida através da acção da enzima viral denominada transcriptase reversa. Assim, o mRNA funciona como molde para a síntese de uma cadeia de DNA. Após a formação da cadeia de DNA, a enzima DNA polimerase actua, formando, por complementaridade, a outra cadeia de DNA a partir dos nucleótidos presentes no meio, constituindo-se uma molécula estável.
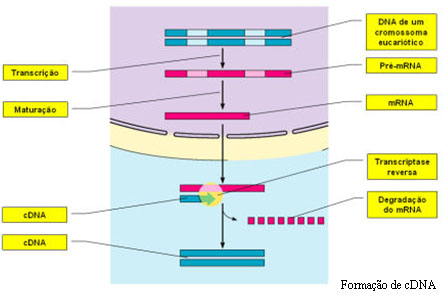
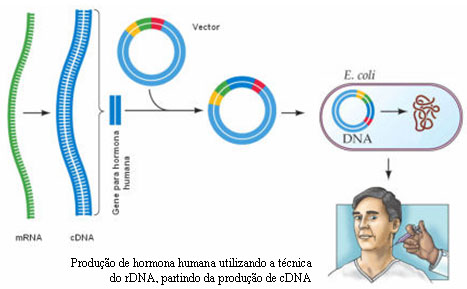
Os pesquisadores recorrem a esta técnica quando se torna necessário clonar genes sem os seus intrões. É de referir que as bactérias utilizadas, por exemplo, na técnica do DNA recombinante, por serem seres procariontes, não têm mecanismos de maturação do DNA e, desta forma, quando se incorporam genes com intrões nestes seres, eles executam a sua transcrição ininterruptamente, incluindo os intrões, gerando uma proteína diferente da desejada. Assim, é inserido um clone de cDNA, de modo a garantir a produção correcta da proteína.
Para além disto, a comparação entre a molécula de cDNA, que não possui intrões, com a molécula de DNA original permite localizar os exões, ou seja, as regiões que verdadeiramente contribuem para a produção da proteína, e os intrões, ou seja, as regiões não codificantes de um determinado gene.
É importante referir ainda que a descoberta da transcriptase reversa e do seu mecanismo de acção e, consequentemente, da tecnologia do cDNA, abriu um importante caminho na biologia molecular. A molécula de RNA é extremamente instável e, dependendo do RNA em questão, o número de cópias pode estar muito limitado. Desta forma, a possibilidade de se trabalhar com cDNA ao invés de RNA tem facilitado muito o trabalho dos cientistas, já que o cDNA é uma molécula estável, de fácil manuseamento e a sua multiplicação é bastante simples.
Reacção de Polimerização em Cadeia (Polymerase Chain Reaction - PCR)
O processo de PCR foi inventado por Kary Mullis em 1983, tendo-lhe sido atribuído o Prémio Nobel da Química pelo seu trabalho. Este processo baseia-se no processo de replicação de DNA que ocorre in vivo e é uma das técnicas para clonar DNA de modo a obter grandes quantidades a partir de uma pequena amostra.

Este é um método muito sensível de análise e, por isso, é realizado com muito cuidado para evitar contaminações que possam inviabilizar ou falsear o resultado. Em primeiro lugar, deve-se extrair o material genético da célula com interesse sem o danificar. Normalmente, o material extraído é o DNA mas pode-se trabalhar também com o RNA num método designado RT-PCR (reverse transcription polymerase chain reaction) que é uma variante do PCR e possui outras aplicações.
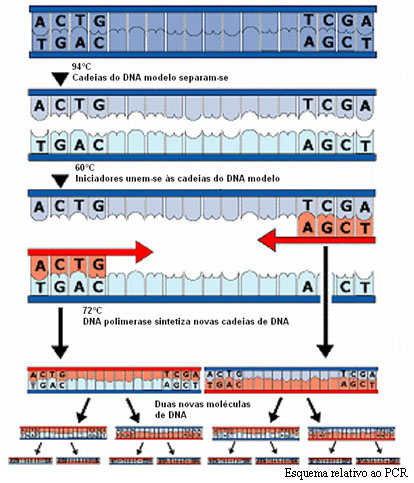
Depois de extraído o DNA, é-lhe adicionada uma mistura que contém nucleótidos (adenina, timina, guanina e citosina), oligonucleótidos iniciadores (primers) e a enzima DNA polimerase. Toda esta mistura é colocada na máquina de PCR, o termo-ciclador, que efectua ciclos de temperatura preestabelecidos com tempos exactos.

Na primeira etapa do ciclo a temperatura é elevada a 94ºC por pouco tempo para que haja a separação da dupla cadeia de DNA a amplificar. Na segunda etapa a temperatura desce para próximo dos 60ºC para que os iniciadores, segmentos de DNA em cadeia simples e geralmente constituídos por 15 a 30 nucleótidos, obtidos por síntese química, se emparelhem com o DNA. Para amplificar uma determinada região são necessários dois iniciadores complementares das sequências que se encontram no início do fragmento de DNA a amplificar, nos seus terminais 3´, de modo a permitir a actuação da DNA polimerase durante a síntese da cadeia complementar. Na última etapa do ciclo, a temperatura é elevada a 72ºC para que a DNA polimerase possa actuar, sintetizando a nova molécula a partir de cada uma das duas cadeias simples constituintes do DNA a amplificar.
De seguida, inicia-se um novo ciclo. Normalmente são realizados de 25 a 30 ciclos para cada reacção, resultando num número de cópias igual a 2nº de ciclos.
O resultado é visualizado e analisado através de uma electroforese (de que falaremos posteriormente) em gel de agarose.
Entre as principais técnicas resultantes de modificações do PCR podemos citar:
- RT-PCR Utiliza a transcriptase reversa para converter uma amostra de RNA em cDNA antes da etapa de amplificação por PCR;
- Nested PCR Para melhorar a especificidade e a eficiência do PCR, o segmento de DNA é amplificado primeiro de forma abrangente, e depois, utilizando este primeiro produto, efectua-se a amplificação da real sequência de nucleótidos do DNA que se pretende;
- PCR multiplex Mais de um segmento de DNA com interesse é amplificado numa única reacção, cada um com seu par de iniciadores específico;
- PCR competitiva Além do DNA molde, é adicionado à reacção um outro segmento de DNA, de sequência, tamanho e concentração conhecidos (controlo), cujas extremidades são complementares também aos iniciadores que irão amplificar a sequência-alvo. O resultado é a amplificação de dois segmentos de DNA. O segmento de controlo, tendo em conta a quantidade inicial e dados sobre a eficácia da reacção serve de padrão para a quantificação do DNA-alvo. Assim, conhecendo a quantidade final do fragmento de controlo e as condições da reacção, podemos dizer a quantidade de DNA-alvo que foi amplificado;
- PCR em tempo real Permite que a amplificação e a quantificação do DNA ocorram simultaneamente, num sistema fechado, sendo necessário um termo-ciclador especializado.
O segredo do sucesso do PCR reside na capacidade que tem de amplificar uma sequência precisa de DNA, aliada à sua simplicidade, rigor, elevada sensibilidade, especificidade e, ainda, baixo custo. Não é necessário isolar o DNA que se pretende amplificar (mesmo que se encontre misturado com DNA de outras espécies), uma vez que a especificidade da PCR é dada pelos iniciadores. É uma técnica rápida, barata e segura.
Contudo, a PCR também tem limitações, como a necessidade de conhecer a sequência de DNA a amplificar para que possam ser sintetizados iniciadores específicos, a relativa facilidade com que ocorre contaminação da amostra por DNA estranho e a limitada extensão da sequência que é possível amplificar. Pode também ocorrer incorporação errada de bases durante a replicação.
O PCR é principalmente utilizado em situações em que a quantidade de DNA disponível é reduzida. Uma das principais aplicações do PCR é na medicina forense, em que pequenas amostras de DNA retiradas da cena de um crime são amplificadas para serem analisadas pelo método de fingerprinting.
Os resultados da PCR são úteis no diagnóstico e prognóstico de doenças. A PCR é frequentemente utilizada na detecção de mutações pontuais e de infecção por microrganismos bacterianos ou virais.
Outro exemplo de aplicação da PCR é no DPI (diagnóstico pré-implantatório) e no DPN (diagnóstico pré-natal). A PCR pode ser usada em DPN em etapas precoces da gestação, permitindo, por exemplo, a detecção de células de um feto em circulação no sangue materno e averiguar a compatibilidade entre o grupo sanguíneo da mãe e do feto. Permite ainda a detecção de várias anomalias cromossómicas como a trissomia 21.
Impressões Digitais Genéticas ou DNA fingerprinting
A estrutura do DNA de todos os seres humanos é a mesma, a única diferença entre as pessoas (e qualquer animal) é a ordem dos nucleótidos. Há tantos milhões de nucleótidos no DNA de cada ser humano que cada pessoa tem uma sequência diferente.
Usando somente estas sequências, cada pessoa pode ser identificada. Contudo, devido à existência de milhões de nucleótidos, a tarefa seria morosa. Em vez disso, os cientistas usam um método mais rápido, que se baseia na existência de padrões repetitivos no DNA, denominados, na ciência forense, de DNA minissatélites. Os cientistas seleccionam determinadas enzimas de restrição, que dividem o DNA nestes segmentos de restrição cujas dimensões, composição em nucleótidos e localização variam muito de pessoa para pessoa e reflectem as diferenças entre os alelos dos vários loci.
Diferentes fragmentos de DNA movimentam-se de modo diferente quando submetidos a electroforese. Esta técnica, também denominada separação electroforética, baseia-se na migração de moléculas carregadas por aplicação de um campo eléctrico. Este processo é frequentemente usado para separar e algumas vezes purificar macromoléculas, especialmente proteínas e ácidos nucleicos, com base no seu tamanho, carga eléctrica ou conformação. A electroforese de ácidos nucleicos e proteínas é realizada normalmente em géis de agarose ou de poliacrilamida. Apesar dos géis de poliacrilamida permitirem uma maior resolução, são os géis de agarose, não tóxicos, os escolhidos para a separação de ácidos nucleicos. Os géis de poliacrilamida, com maior resolução mas com menor gama de separação que os géis de agarose, são normalmente usados na separação e caracterização de misturas de proteínas.

A electroforese de DNA é realizada em gel de agarose. Quando sujeitos a um campo eléctrico, os ácidos nucleicos migram em direcção ao pólo positivo, uma vez que apresentam carga negativa. A agarose funciona como uma rede cujos poros deixam passar mais facilmente as moléculas de menor tamanho, que vão, portanto, migrar mais do que as de maiores dimensões. A migração de um fragmento de DNA na forma circular, como um plasmídeo não digerido, é diferente da migração do mesmo plasmídeo sob a forma linear (após digestão com uma enzima de restrição). Por outro lado, moléculas com uma configuração mais compacta migram mais rapidamente do que moléculas com uma conformação molecular mais distendida.
A electroforese em gel é frequentemente utilizada para estimar o tamanho de fragmentos de ácidos nucleicos. Através da comparação da distância percorrida pelos fragmentos com a percorrida por fragmentos de peso molecular conhecido (padrões de peso molecular) é possível inferir sobre o peso molecular de cada fragmento da amostra a analisar.
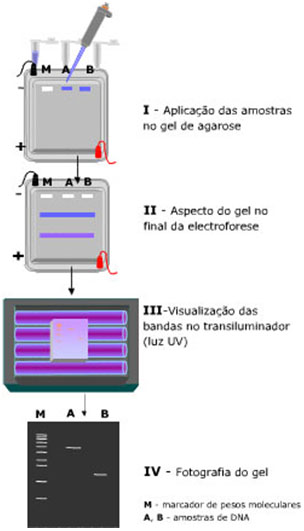
Posto isto, para executar a técnica das impressões digitais genéticas, é necessário que, em primeiro lugar, o DNA, submetido à acção de enzimas de restrição, se fragmente em porções de diferentes tamanhos e pesos moleculares.
Depois, prepara-se o gel de agarose que é imerso numa tina de electroforese com uma solução-tampão que estabelece a condução eléctrica com a fonte de alimentação.
Uma vez aplicadas as amostras de DNA e o padrão de pesos moleculares nos poços, inicia-se a electroforese. A aplicação do campo eléctrico é efectuada através de 2 eléctrodos situados paralelamente à fileira de poços.
Terminada a separação electroforética, o gel deve ser imerso numa solução de brometo de etídeo que permitirá a visualização das bandas de DNA por exposição a luz ultravioleta.
O padrão de fragmentos de restrição é único de indivíduo para indivíduo, funcionando como um código de barras genético.
As principais aplicações da técnica do DNA fingerprinting são as seguintes:
- Genética forense A dactiloscopia genética é uma técnica utilizada na ciência forense onde se analisam as diferenças entre os DNA minissatélites, já referidos, provenientes de material biológico deixado num local (sangue, cabelo, esperma
). Estes segmentos encontram-se entre os genes. A hipótese de dois indivíduos sem qualquer relação de parentesco possuírem exactamente os mesmos minissatélites é muito remota, por isso a dactiloscopia do DNA constitui um teste viável. A região minissatélite é cortada em fragmentos por enzimas de restrição que são separados através de electroforese. É produzido um padrão de bandas de DNA. Os padrões de indivíduos diferentes podem ser comparados e as diferenças são facilmente detectadas e as pessoas distinguidas. Isto permite a identificação de cadáveres e a identificação de criminosos. Contudo, na análise dos suspeitos de um crime, por exemplo, é de ter em conta se estes têm irmãos gémeos, uma vez que, através das impressões digitais genéticas, não se podem distinguir dois gémeos monozigóticos, pois os padrões de banda são os mesmos.
- Determinação de paternidade Como cada indivíduo herda a disposição dos seus nucleótidos dos seus pais, comparando os padrões de banda de um indivíduo com os dos alegados progenitores, podem-se obter probabilidades de parentesco que nos levem a excluir a paternidade ou a confirmá-la com um elevado grau de certeza. Se os dois padrões forem suficientemente semelhantes (tendo em conta que só metade do DNA é herdado de cada progenitor), então a paternidade confirma-se.
- Se utilizada em conjunto com metodologias sociológicas, a técnica das impressões digitais genéticas pode ser usada para analisar padrões de migração e confirmar origens de étnicas.
- Esta tecnologia pode ser usada para prever a nossa saúde futura. Assim, pode ser utilizada para localizar os genes de doenças hereditárias. Se um padrão particular de DNA aparece repetidamente em diferentes doentes, os cientistas podem verificar qual(is) o(s) gene(s), ou pelo menos qual(is) pedaço(s) de DNA, que poderá(ão) estar envolvido(s). Como o conhecimento dos genes envolvidos na susceptibilidade a uma doença dá pistas sobre fisiologia subjacente à doença, o DNA fingerprinting auxilia no desenvolvimento de terapias. Pode também ser usado no período pré-natal para detectar possíveis anomalias hereditárias, como a distrofia muscular ou a doença de Huntington, de forma a que possa ser disponibilizado aconselhamento médico e possam ser adoptadas as devidas precauções.
|
|

