
Total de visitas: 337843
|
Hereditariedade e Doenças Genéticas
Hereditariedade A Base Mendeliana

As bases da hereditariedade foram propostas há mais de um século por um padre católico e professor chamado Mendel. A partir das suas experiências com ervilheiras de cheiro Mendel foi capaz de reconhecer os princípios fundamentais da hereditariedade que hoje a comunidade científica aplica a quase todos os organismos que se reproduzem sexuadamente. Porém, e como muitas vezes se verificou ao longo de séculos de construção de conhecimento científico, apesar do cuidado da sua interpretação e da minúcia e objectividade com que apresentou o resultado do seu trabalho aos biólogos da época, as suas pesquisas e consequentes conclusões permaneceram na escuridão durante quase três décadas. Muitos estudiosos modernos têm tentado perceber o porquê da não aceitação de um trabalho tão rico e importante e segundo se tem concluído, como várias vezes aconteceu ao longo da história, o cientista era demasiado avançado para a época em que vivia. Lembremo-nos, por exemplo, de Albert Einstein, cujas descobertas só foram comprovadas cerca de meio século após a sua morte.
Nas três décadas que se seguiram à contribuição de Mendel, passos gigantes foram dados na compreensão da natureza das células e da reprodução celular. Acontecimentos verdadeiramente históricos tiveram lugar nesta fase e marcaram de forma latente a comunidade científica: a divisão celular e a fecundação foram observadas, foram descobertos os cromossomas e descritos os seus movimentos nos processos mitóticos e meióticos, e a substância química do núcleo foi isolada e caracterizada. Deste modo no início do século XX prevalecia já a ideia, ainda que apenas por parte de alguns notáveis biólogos, que o material genético estava localizado no núcleo celular e não armazenado em proteínas como até há bem pouco tempo se suponha ser verdade. Por esta altura, e no decorrer dos seus próprios estudos, vários investigadores foram levados à redescoberta dos princípios que Mendel havia enunciado vários anos antes. Perante estas novas perspectivas a obra de Mendel foi reavaliada tendo sido reconhecida a sua importância fundamental. Pouco tempo depois, a comparação entre o comportamento dos cromossomas e as unidades de hereditariedade postuladas por Mendel denotou uma analogia perfeita tendo sido finalmente identificadas as estruturas onde estavam localizadas as unidades hereditárias. Como forma de auxílio à redacção deste texto foi utilizada a seguinte fonte literária: JONES, Kenneth; GAUDIN, Anthon J. - Introdução à Biologia 3ª edição (1977). Lisboa: Fundação Calouste Gulbenkian.
A Base Cromossómica da Hereditariedade
Os genes são unidades hereditárias que contêm a informação para a produção de substâncias bioquímicas específicas na célula. Os genes estão dispostos linearmente nos cromossomas ao longo do núcleo celular. A meiose é um processo de divisão celular que leva à formação de gâmetas. Durante a meiose o número de cromossomas é reduzido a metade, sendo que é na fecundação que o número total de cromossomas característico de cada espécie é restabelecido através da união do oócito II (gâmeta sexual feminino) com o espermatozóide (gâmeta sexual masculino) originando-se um zigoto que possui informação genética recebida por via materna e informação genética recebida por via paterna. Os cromossomas idênticos em diferentes guarnições dizem-se homólogos. Eles possuem os mesmos genes e uma estrutura aparentemente semelhante.
Doenças Genéticas, afinal o que são?
Os genes são a informação bioquímica herdada dos nossos pais no momento da concepção, como aliás anteriormente referi. Determinam a nossa constituição biológica. São eles que nos tornam parecidos com os nossos progenitores e controlam o nosso crescimento e a nossa aparência. Também determinam a nossa resistência a determinadas doenças assim como a predisposição que possuímos em relação a outras. As chamadas doenças genéticas.
Mais à frente neste trabalho irei abordar os mecanismos pelos quais os defeitos genéticos são transmitidos de geração em geração.
Pequena Introdução sobre a Temática das Doenças Genéticas
Durante as últimas décadas, foram descobertos na espécie humana vários genes que existem em duas ou mais formas alélicas (formas alternativas de um mesmo gene). Em muitos casos, as diferenças no efeito provocado pelas diferentes formas são relativamente inócuas, uma vez que não constituem nem uma vantagem nem uma desvantagem adaptativa para os indivíduos. Porém, em muitos outros casos, a forma alélica de um gene está intimamente relacionada com uma doença genética ou então com uma malformação ao nível do desenvolvimento, pelo que assume grande relevância o facto dos indivíduos terem conhecimento da sua própria ascendência genética e da do seu potencial companheiro. Este conhecimento irá permitir-lhes que calculem inteligentemente a probabilidade de um carácter, à partida não desejável, aparecer nos seus filhos, ao mesmo tempo que os irá preparar para a eventualidade de terem de enfrentar o efeito de uma doença genética. Um óptimo exemplo de como tal prevenção pode ser útil relaciona-se com uma patologia denominada galactosemia. Os indivíduos afectados por esta doença são homozigóticos (indivíduos que apresentam um genótipo formado por dois alelos iguais para determinado carácter) em relação a um gene recessivo, cuja forma dominante, normal, é responsável por uma enzima utilizada para metabolizar o açúcar e o leite. Estes indivíduos produzem uma enzima deficiente e são incapazes de utilizar a lactose, o açúcar vulgar que se encontra no leite. Os efeitos desta patologia começam a sentir-se bastante cedo, por volta de uma semana após a ocorrência do nascimento. É bastante provável, perante a ausência de um diagnóstico convenientemente precoce, que a criança seja atacada por icterícia, tenha diarreia e sofra de graves diminuições das funções do fígado e dos rins. Em casos mais extremos, a morte segue-se um pouco mais tarde. Nos casos em que a criança sobrevive possui um crescimento atrofiado e um atraso mental acentuado. Ora acontece que se o diagnóstico for suficientemente precoce, o desenvolvimento físico e mental pode perfeitamente ser normal através unicamente da eliminação da lactose da alimentação, fornecendo à criança açúcar sob outras formas.
Contudo outros casos podem ser mais sérios e mesmo incuráveis. São conhecidas várias doenças debilitantes devidas a causas genéticas específicas. Estas incluem a distrofia muscular, a diabetes, a hemofilia, a anemia falciforme e a doença de Tay-Sach, isto nomeando apenas algumas. No último caso, um embrião pode ser testado logo no início do seu desenvolvimento procedendo-se à extracção de uma pequena quantidade de líquido do âmnio, o saco de líquido que envolve o embrião. Deste modo, uma criança que possua esta patologia, poderá obter um diagnóstico bastante satisfatório ainda antes do próprio nascimento. Com o estado actual da tecnologia médica, a criança nascerá, viverá alguns anos, perderá gradualmente o controlo muscular e a coordenação nervosa por deterioração progressiva do sistema nervoso central, acabando por morrer. Os pais, alertados para a possibilidade do seu filho vir a possuir esta anomalia genética, podem descobrir no início da gravidez da mãe que a criança será afectada, e, eventualmente, decidir-se pela interrupção da gravidez.
As doenças genéticas podem igualmente ser provocadas por alelos dominantes que, ao contrário dos recessivos, possuem a capacidade de se manifestar nos indivíduos heterozigóticos (indivíduos que apresentam um genótipo formado por dois alelos diferentes). Uma doença chamada aplasia da retina, por exemplo, causa a cegueira no homem e é herdada como um gene dominante. Os indivíduos que são heterozigóticos em relação a este gene e ao seu alelo recessivo normal sofrem de uma malformação da retina que acaba por lhes provocar a cegueira.
É oportuno esclarecer nesta fundamentação teórica uma dúvida que nos surge ao analisar vários tipos de livros relacionados directamente com esta temática das doenças genéticas: um alelo dominante não é necessariamente a forma mais comum de um gene. A verdade é que se conhecem vários casos onde um alelo dominante é extremamente raro numa população, e a forma mais comum é um recessivo em relação a ele.
A anemia de células falciformes ou siclemia é uma doença relativamente vulgar nas regiões do velho mundo, sendo especialmente predominante em certas regiões da África, da Índia meridional e em algumas regiões dos países limítrofes do mar Mediterrâneo. Os indivíduos que são homozigóticos em relação ao gene que provoca esta doença sofrem de atraso no desenvolvimento, elevadas taxas de destruição de sangue durante as infecções, ulcerações e outros problemas. A morte ocorre algumas vezes na infância, mas por vezes as pessoas afectadas sobrevivem até serem adultas. A doença foi assim denominada devido ao aspecto dos glóbulos vermelhos quando os tecidos contêm baixos níveis de oxigénio. O glóbulo vermelho encurva-se e aparece em forma de foice quando visto de lado, e não achatado como os glóbulos normais.
Uma forma mais moderna da doença manifesta-se em indivíduos que são heterozigóticos para o gene da siclemia, anomalia essa que só raramente ocasiona uma incapacidade. Os glóbulos dos indivíduos heterozigóticos apresentam igualmente uma forma de foice, tendo este acontecimento apenas lugar perante níveis de oxigénio bastante baixos.
Na espécie humana, o estudo da transmissão de características hereditárias, não pode ser feito de forma tão prática como aconteceu com Mendel, que se serviu das suas ervilheiras de cheiro fazendo delas o material biológico da sua pesquisa. Aos estudos sobre esta temática incidentes na espécie humana colocam-se vários entraves como o facto do tempo de uma geração ser muito longo, o número de descendentes por geração ser bastante baixo, não se efectuarem cruzamentos experimentais e sermos uma espécie com um elevado número de cromossomas.
Pelas razões supracitadas, os estudos de hereditariedade humana baseiam-se principalmente na análise de árvores genealógicas.
Uma árvore genealógica, ou pedigree, é um esquema que permite seguir a transmissão de certos caracteres numa família ao longo de várias gerações. A análise de uma árvore genealógica reveste-se de uma crucial importância na determinação da origem de certas doenças ou anomalias, permitindo igualmente inferir os riscos da sua transmissão às gerações vindouras. No âmbito deste trabalho apresentar-se-ão mais à frente alguns exemplos de árvores genealógicas.
Hereditariedade Autossómica
A transmissão de caracteres hereditários codificados por genes que se localizam nos autossomas reveste-se de aspectos particulares, conforme o alelo responsável pela manifestação do carácter é dominante ou recessivo. De seguida iremos expor os aspectos particulares da transmissão de caracteres dominantes e recessivos, acompanhando cada um dos modos de transmissão com alguns exemplos de doenças genéticas que lhe estão vedadas.
Transmissão Autossómica Dominante:
Neste tipo de transmissão mulheres e homens são afectados pelas patologias que lhe estão subjacentes com a mesma frequência, transmitindo igualmente um carácter com a mesma frequência. Desta forma, sucessivas gerações são afectadas, sendo que a transmissão do carácter apenas pára numa geração em que não se verifica a presença de um único indivíduo afectado. Pelo menos um dos progenitores de um indivíduo afectado pela anomalia também é afectado. Os indivíduos afectados são homozigóticos dominantes ou heterozigóticos.
Um dos exemplos de patologias ligadas a este tipo de transmissão é a polidactilia. Esta anomalia caracteriza-se pelo aparecimento de mais que 5 dedos nas mãos ou nos pés. Outra das anomalias dá pelo nome de hipercolesterolémia familiar. Os indivíduos portadores desta patologia têm níveis altos de colesterol desde o dia do seu nascimento, havendo, por isso, um risco consideravelmente acrescido de doenças cardiovasculares prematuras.
Manifestações fenotípicas igualmente associadas a este tipo de transmissão de características são: orelhas de lóbulo solto, capacidade de enrolar a língua, cabelo castanho e visão normal.
Transmissão Autossómica Recessiva:
Similarmente ao que se verifica no modo de transmissão autossómica dominante, também neste tipo de transmissão são afectados com a mesma frequência indivíduos do sexo masculino e indivíduos do sexo feminino, transmitindo o carácter com a mesma frequência. A transmissão do carácter pode saltar gerações sendo geralmente mais difícil de identificar que os caracteres associados a um modo de transmissão dominante.
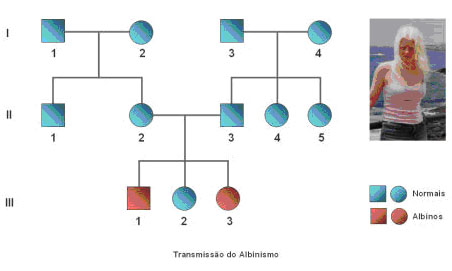
Os pais de um indivíduo afectado pela patologia ou manifestam o carácter ou são obrigatoriamente portadores, sendo que o indivíduo afectado é sempre um homozigótico recessivo. Os heterozigóticos são portadores.
Como exemplos de doenças associadas a este tipo de transmissão de características temos, nomeando apenas alguns, o albinismo, a fibrose quística, a fenilcetonúria e a surdez. O albinismo é uma doença pouco frequente e hereditária caracterizada pela incapacidade de formação de melanina. As pessoas com albinismo (albinos) podem ter o cabelo branco, a pele pálida e os olhos rosados. Podem também apresentar, com frequência, visão anormal e movimentos oculares involuntários. Devido ao facto de a melanina proteger a pele da acção do sol, os albinos são muito propensos às queimaduras solares e, consequentemente, aos cancros da pele. A fibrose quística é uma doença hereditária que faz com que certas glândulas produzam secreções anormais, cujo resultado é uma série de sintomas, entre os quais o mais importante afecta o tubo digestivo e os pulmões. Esta doença é de elevada mortalidade, sendo que grande parte dos seus portadores morrem jovens e apenas 35% chegam à idade adulta. No entanto, a sua incidência é desigual nas diferentes etnias; assim, nos Estados Unidos, por exemplo, onde a diversidade étnica e a qualidade dos estudos estatísticos fornecem dados comparativos, a fibrose quística, que é a causa mórbida de maior frequência entre as pessoas de raça branca, sendo 5 % portadores de um gene anormal portador da doença, causa a morte de 1 em cada 2500 bebés de raça branca e de 1 em cada 17 000 de raça negra, enquanto é rara entre os indivíduos asiáticos. Nas pessoas que são portadoras do gene anormal responsável pela referida patologia, a doença, uma vez que o traço é recessivo, manifesta-se unicamente quando são dois os genes anormais que o indivíduo possui. Os sintomas são totalmente imperceptíveis em pessoas portadoras de um só gene anormal. Este gene controla a produção de uma proteína que regula a passagem de cloro e sódio (o corrente sal) através das membranas celulares. Quando ambos os genes são anormais, esta passagem interrompe-se, provocando desidratação e aumento da viscosidade das secreções. A fibrose quística afecta a quase totalidade das glândulas exócrinas (glândulas que segregam líquidos para o interior de um canal). As secreções são anormais e afectam o funcionamento glandular. Em algumas glândulas, como o pâncreas e as glândulas dos intestinos, as secreções são espessas ou sólidas e podem obstruir completamente a glândula. As glândulas que produzem muco nas vias aéreas dos pulmões fabricam secreções anormais que as obstruem, permitindo a multiplicação de bactérias. As glândulas sudoríparas, as glândulas parótidas e as pequenas glândulas salivares segregam líquidos cujo conteúdo em sal é superior ao normal. A fenilcetonúria (FCU, fenilalaninemia ou oligofrenia fenilpirúvica) é um problema hereditário no qual a enzima que processa o aminoácido fenilalanina se encontra ausente, provocando um valor alarmantemente alto de fenilalanina no sangue. A fenilalanina transforma-se normalmente em tirosina, outro aminoácido, eliminando-se do organismo. Sem a enzima que a transforma, a fenilalanina acumula-se no sangue e é tóxica para o cérebro, provocando atraso mental. A fenilalanina está presente na maioria dos grupos geográficos, mas é rara na população judaica de descendentes europeus orientais e na negra. A incidência é de aproximadamente 1 em cada 16 000 nascidos vivos. Por seu lado, a perda de audição define-se precisamente como sendo uma deterioração desta mesma função. A surdez não é mais que uma perda auditiva profunda. A perda da audição pode ser causada por um problema mecânico no canal auditivo ou no ouvido médio que obstrói a condução do som (perda condutiva de audição) ou por uma lesão no ouvido interno, no nervo auditivo ou nas vias do nervo auditivo no cérebro (perda neuro-sensorial da audição). Os dois tipos de perda da audição podem ser diferenciados comparando como uma pessoa ouve os sons conduzidos pelo ar e como os ouve conduzidos pelos ossos.
As orelhas com lóbulo aderente, a inaptidão para enrolar a língua e o cabelo louro são características associadas ao meio de transmissão supracitado.
Hereditariedade ligada ao sexo
As experiências realizadas por Thomas Morgan com a Drosophila melanogaster contribuíram de forma irrefutável para consolidar a teoria cromossómica da hereditariedade revelando igualmente a ligação ao sexo da transmissão de certos caracteres hereditários.
Na espécie humana, o sexo feminino (XX) ou masculino (XY) é determinado no momento da fecundação pela união dos cromossomas transportados pelos gâmetas. Os gâmetas femininos transportam apenas o cromossoma X e, por isso, as mulheres são homogaméticas. Metade dos gâmetas masculinos transportam o cromossoma X e a outra metade transporta o cromossoma Y e, por isso, os homens são heterogaméticos.
Os genes presentes no cromossoma Y são transmitidos de pai para filho e os genes presentes no cromossoma X são transmitidos de pai para filha e de mãe para filho ou filha.
Seguidamente iremos pôr em evidência os aspectos particulares da transmissão de caracteres determinados por alelos dominantes e recessivos, localizados no cromossoma X, seguindo a mesma linha de orientação existente na apresentação dos modos de transmissão relacionados com a hereditariedade autossómica, isto é, os dados relativos a este modo de transmissão irão ser acompanhados com alguns exemplos de tipos de doenças que lhe estão vedadas.
Transmissão de um Alelo Dominante ligado ao Cromossoma X
O carácter em questão exprime-se sempre de uma forma consideravelmente mais severa nos homens do que nas mulheres, sendo que nos indivíduos do sexo feminino o carácter exprime-se em homozigóticos dominantes e em heterozigóticos.
Um homem afectado tem obrigatoriamente uma mãe afectada, enquanto que uma mulher afectada pode ter um pai ou uma mãe afectada.
A síndrome de Rett é um exemplo de uma anomalia relacionada com este tipo de transmissão. Esta patologia é do foro neurológico e ocorre quase exclusivamente em crianças do sexo feminino. Após um período inicial aparentemente normal as crianças afectadas desenvolvem microcefalia com regressão do desenvolvimento e alterações neurológicas e comportamentais características. Os sintomas desta patologia são os mais variados e podem ser, inicialmente, confundidos com outras patologias neurológicas, como o autismo. As raparigas que sofrem desta doença nascem sem qualquer sinal aparente de anormalidade e parecem desenvolver-se normalmente entre os 6 e os 18 meses de vida. Subitamente, porém, estagnam na sua evolução e começam a apresentar sinais de regressão física e neurológica. É nesta fase que começa a surgir um quadro sintomático particular. Os bebés começam a perder a capacidade de usar as mãos, mostram ter problemas de equilíbrio, deixam de falar, podem perder a marcha, ficam mentalmente diminuídos e demonstram sérios problemas de relacionamento com os outros, evitando, por exemplo, o contacto visual. A estereotipia mais característica da síndroma é, no entanto, a que se assemelha a um constante esfregar de mãos e que é o sintoma mais comum. A maior parte destas crianças tem uma esperança média de vida diminuída e morre antes de atingir a idade adulta, apesar de haver casos conhecidos de doentes que chegam aos 50 anos. Foi pela primeira vez descrita em 1966, pelo Dr. Andreas Rett de Viena, Áustria, numa publicação médica alemã, daí o nome da patologia.
Transmissão de um Alelo Recessivo ligado ao cromossoma X
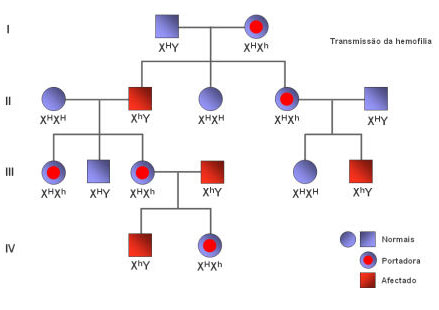
O carácter exprime-se sempre nos indivíduos de sexo masculino, exprimindo-se apenas nas mulheres homozigóticas recessivas. Um homem afectado tem obrigatoriamente uma mãe afectada ou portadora da patologia. Uma mulher afectada tem, obrigatoriamente, um pai afectado e uma mãe afectada ou portadora. A hemofilia, o daltonismo e a própria diabetes são exemplos de anomalias relacionados com o modo de transmissão em questão. A hemofilia é originada pela mutação de um gene responsável pela criação de uma proteína necessária à coagulação sanguínea: Perante a sua inexistência qualquer hemorragia mínima pode durar horas acabando mesmo, em alguns casos, por levar à morte do indivíduo. Simbolicamente, e em árvores genealógicas como a que de seguida se irá apresentar, utiliza-se o h para designar o gene responsável pela hemofilia e o H para designar o gene responsável pela normalidade do indivíduo. O daltonismo, por seu lado, caracteriza-se pela incapacidade de distinguir algumas cores por parte dos indivíduos afectados por esta patologia, com é o caso do vermelho e do verde, ou do cinzento e do castanho. Similarmente à hemofilia, também o modo de transmissão do daltonismo possui uma série de representações simbolicamente quando é transposto para um heredograma. Assim d diz respeito ao alelo portador do daltonismo e o D utiliza-se para identificar os alelos normais. A diabetes caracteriza-se por uma hiperglicémia crónica, ou seja, o nível de açúcar no sangue (glicémia) está aumentado para lá dos valores normais, devido a uma perturbação no funcionamento da hormona da insulina. Normalmente o que a insulina faz é facilitar a entrada de glicose em circulação nas células que a vão utilizar na produção de energia para o seu funcionamento. Se a insulina não existe ou não funciona devidamente, a glicose mantém-se no sangue o que conduz a uma situação de hiperglicémia.
|
|

